 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeApproximation and inference methods for stochastic biochemical kinetics - a tutorial review
Paper and Code
Jan 12, 2017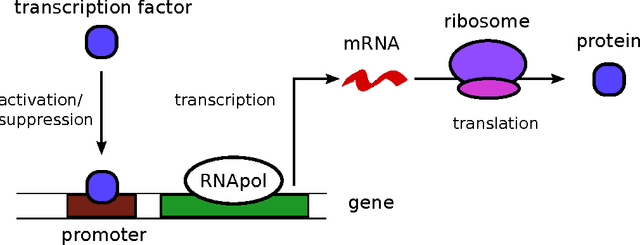
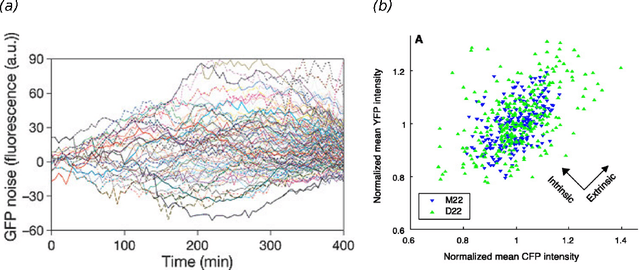


Stochastic fluctuations of molecule numbers are ubiquitous in biological systems. Important examples include gene expression and enzymatic processes in living cells. Such systems are typically modelled as chemical reaction networks whose dynamics are governed by the Chemical Master Equation. Despite its simple structure, no analytic solutions to the Chemical Master Equation are known for most systems. Moreover, stochastic simulations are computationally expensive, making systematic analysis and statistical inference a challenging task. Consequently, significant effort has been spent in recent decades on the development of efficient approximation and inference methods. This article gives an introduction to basic modelling concepts as well as an overview of state of the art methods. First, we motivate and introduce deterministic and stochastic methods for modelling chemical networks, and give an overview of simulation and exact solution methods. Next, we discuss several approximation methods, including the chemical Langevin equation, the system size expansion, moment closure approximations, time-scale separation approximations and hybrid methods. We discuss their various properties and review recent advances and remaining challenges for these methods. We present a comparison of several of these methods by means of a numerical case study and highlight some of their respective advantages and disadvantages. Finally, we discuss the problem of inference from experimental data in the Bayesian framework and review recent methods developed the literature. In summary, this review gives a self-contained introduction to modelling, approximations and inference methods for stochastic chemical kinetics.