 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeAI-driven Hypernetwork of Organic Chemistry: Network Statistics and Applications in Reaction Classification
Paper and Code
Aug 02, 2022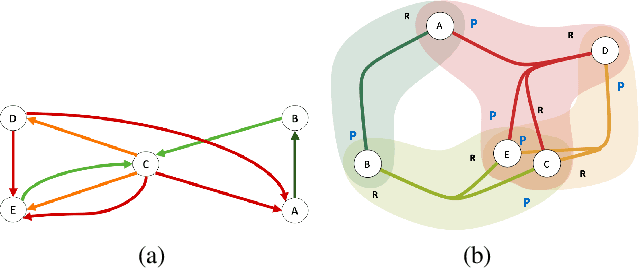
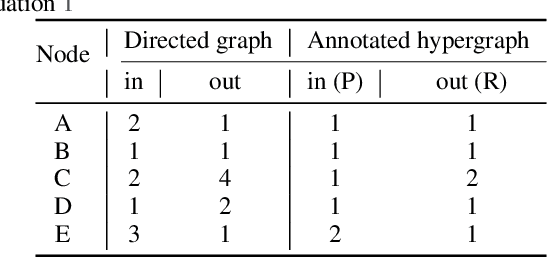
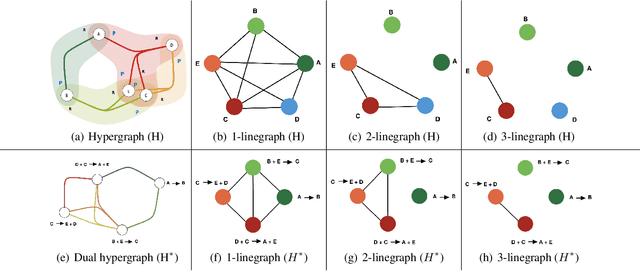
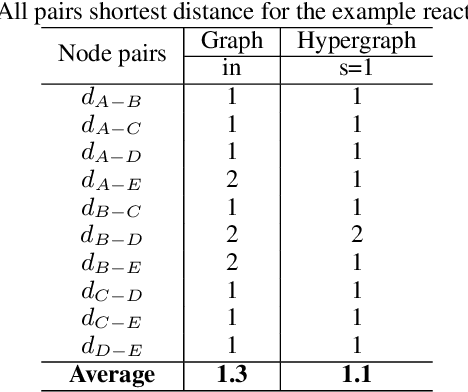
Rapid discovery of new reactions and molecules in recent years has been facilitated by the advancements in high throughput screening, accessibility to a much more complex chemical design space, and the development of accurate molecular modeling frameworks. A holistic study of the growing chemistry literature is, therefore, required that focuses on understanding the recent trends and extrapolating them into possible future trajectories. To this end, several network theory-based studies have been reported that use a directed graph representation of chemical reactions. Here, we perform a study based on representing chemical reactions as hypergraphs where the hyperedges represent chemical reactions and nodes represent the participating molecules. We use a standard reactions dataset to construct a hypernetwork and report its statistics such as degree distributions, average path length, assortativity or degree correlations, PageRank centrality, and graph-based clusters (or communities). We also compute each statistic for an equivalent directed graph representation of reactions to draw parallels and highlight differences between the two. To demonstrate the AI applicability of hypergraph reaction representation, we generate dense hypergraph embeddings and use them in the reaction classification problem. We conclude that the hypernetwork representation is flexible, preserves reaction context, and uncovers hidden insights that are otherwise not apparent in a traditional directed graph representation of chemical reactions.