 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeAccelerating the discovery of low-energy structure configurations: a computational approach that integrates first-principles calculations, Monte Carlo sampling, and Machine Learning
Paper and Code
Oct 08, 2024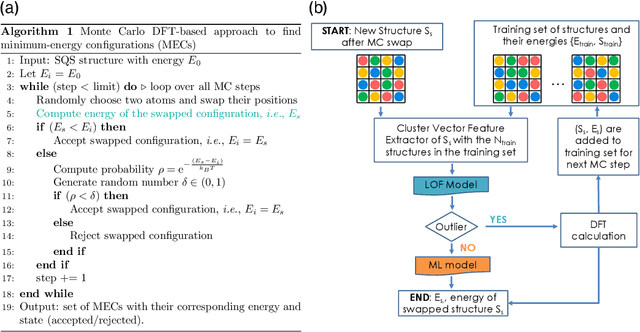
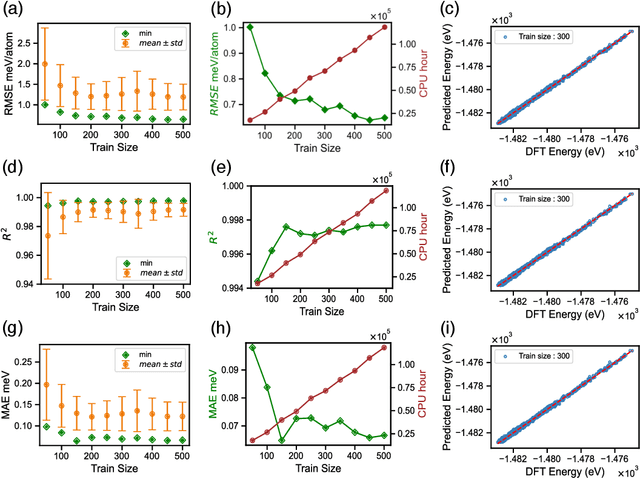
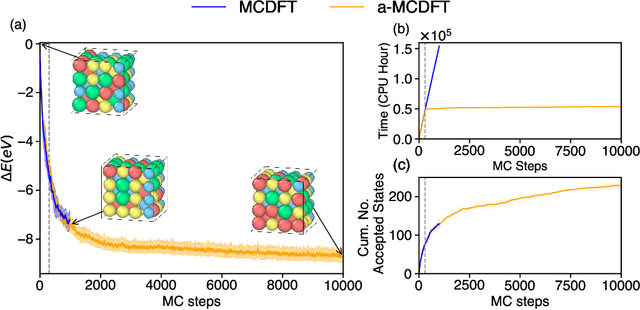
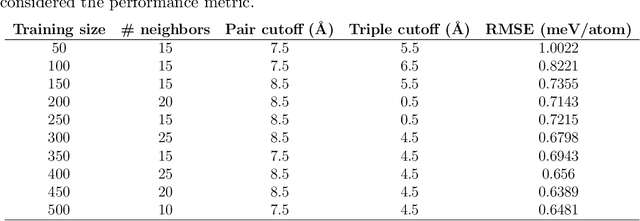
Finding Minimum Energy Configurations (MECs) is essential in fields such as physics, chemistry, and materials science, as they represent the most stable states of the systems. In particular, identifying such MECs in multi-component alloys considered candidate PFMs is key because it determines the most stable arrangement of atoms within the alloy, directly influencing its phase stability, structural integrity, and thermo-mechanical properties. However, since the search space grows exponentially with the number of atoms considered, obtaining such MECs using computationally expensive first-principles DFT calculations often results in a cumbersome task. To escape the above compromise between physical fidelity and computational efficiency, we have developed a novel physics-based data-driven approach that combines Monte Carlo sampling, first-principles DFT calculations, and Machine Learning to accelerate the discovery of MECs in multi-component alloys. More specifically, we have leveraged well-established Cluster Expansion (CE) techniques with Local Outlier Factor models to establish strategies that enhance the reliability of the CE method. In this work, we demonstrated the capabilities of the proposed approach for the particular case of a tungsten-based quaternary high-entropy alloy. However, the method is applicable to other types of alloys and enables a wide range of applications.