 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeLearning local and semi-local density functionals from exact exchange-correlation potentials and energies
Sep 10, 2024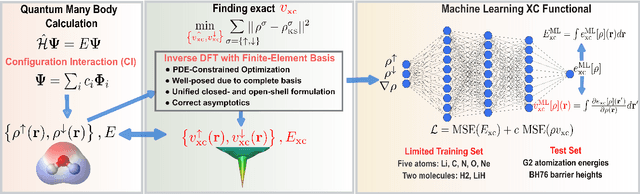



Finding accurate exchange-correlation (XC) functionals remains the defining challenge in density functional theory (DFT). Despite 40 years of active development, the desired chemical accuracy is still elusive with existing functionals. We present a data-driven pathway to learn the XC functionals by utilizing the exact density, XC energy, and XC potential. While the exact densities are obtained from accurate configuration interaction (CI), the exact XC energies and XC potentials are obtained via inverse DFT calculations on the CI densities. We demonstrate how simple neural network (NN) based local density approximation (LDA) and generalized gradient approximation (GGA), trained on just five atoms and two molecules, provide remarkable improvement in total energies, densities, atomization energies, and barrier heights for hundreds of molecules outside the training set. Particularly, the NN-based GGA functional attains similar accuracy as the higher rung SCAN meta-GGA, highlighting the promise of using the XC potential in modeling XC functionals. We expect this approach to pave the way for systematic learning of increasingly accurate and sophisticated XC functionals.