 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeDe novo design of protein target specific scaffold-based Inhibitors via Reinforcement Learning
May 21, 2022
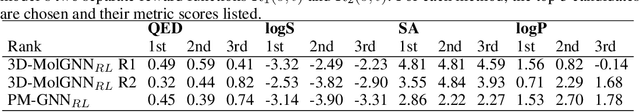

Efficient design and discovery of target-driven molecules is a critical step in facilitating lead optimization in drug discovery. Current approaches to develop molecules for a target protein are intuition-driven, hampered by slow iterative design-test cycles due to computational challenges in utilizing 3D structural data, and ultimately limited by the expertise of the chemist - leading to bottlenecks in molecular design. In this contribution, we propose a novel framework, called 3D-MolGNN$_{RL}$, coupling reinforcement learning (RL) to a deep generative model based on 3D-Scaffold to generate target candidates specific to a protein building up atom by atom from the starting core scaffold. 3D-MolGNN$_{RL}$ provides an efficient way to optimize key features by multi-objective reward function within a protein pocket using parallel graph neural network models. The agent learns to build molecules in 3D space while optimizing the activity, binding affinity, potency, and synthetic accessibility of the candidates generated for infectious disease protein targets. Our approach can serve as an interpretable artificial intelligence (AI) tool for lead optimization with optimized activity, potency, and biophysical properties.