 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgePrediction of transport property via machine learning molecular movements
Mar 07, 2022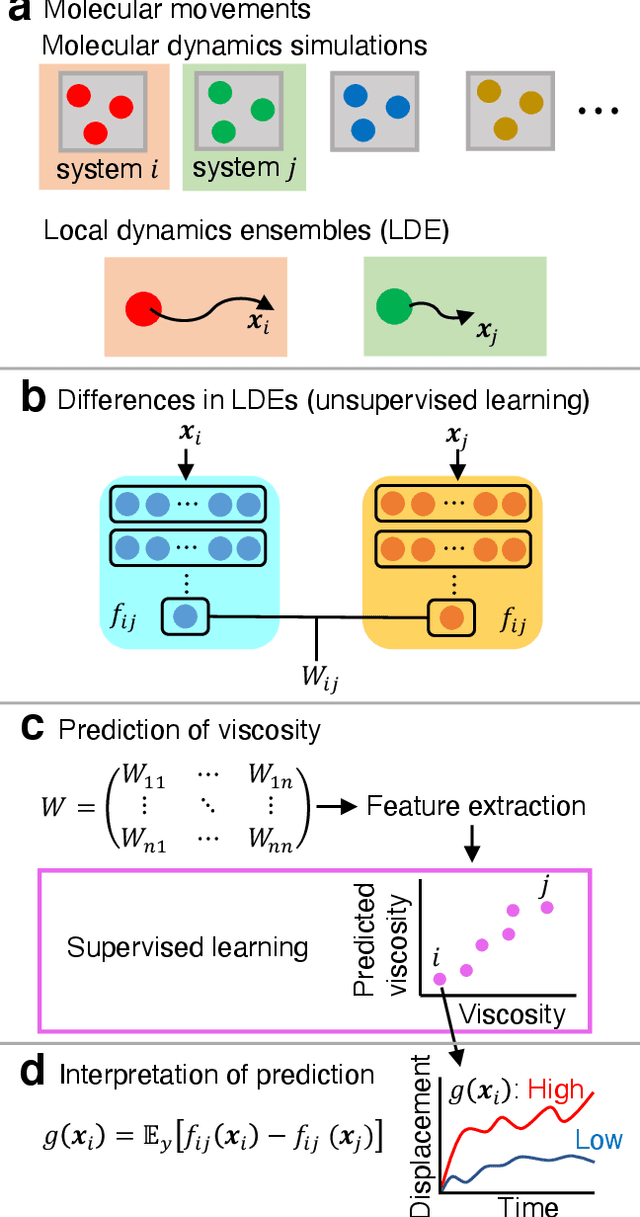
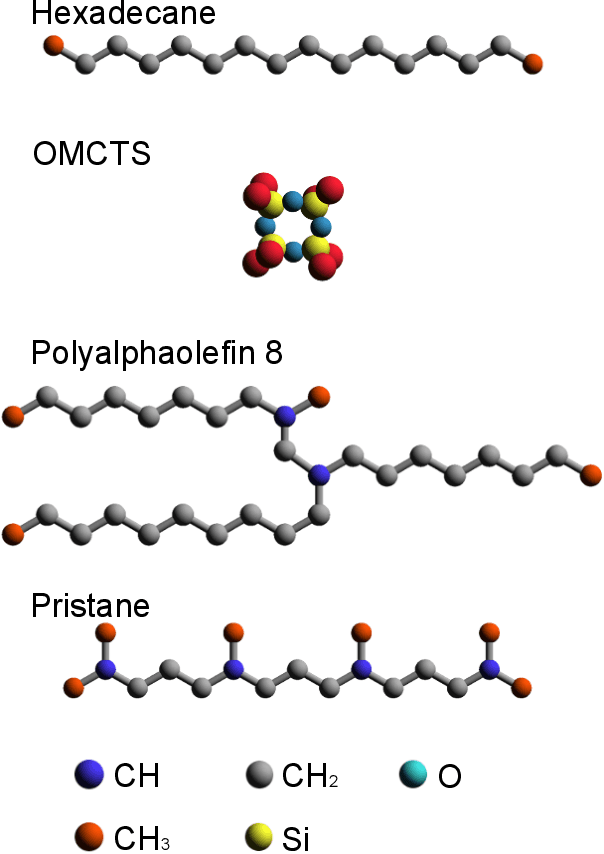
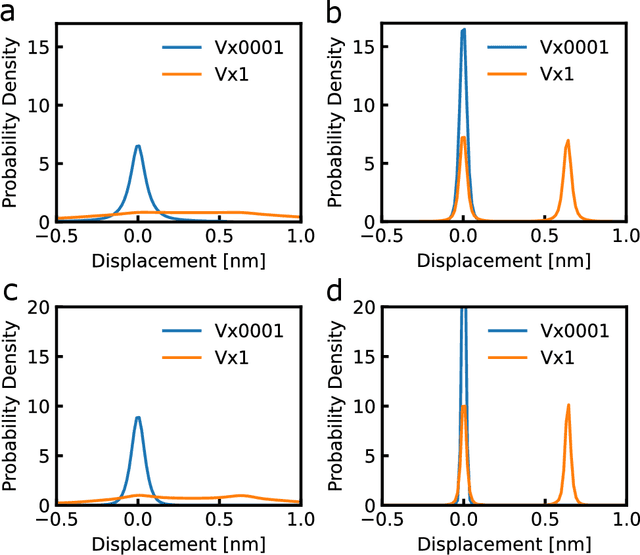
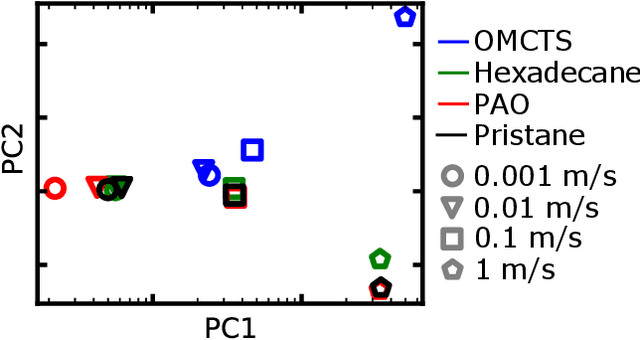
Molecular dynamics (MD) simulations are increasingly being combined with machine learning (ML) to predict material properties. The molecular configurations obtained from MD are represented by multiple features, such as thermodynamic properties, and are used as the ML input. However, to accurately find the input--output patterns, ML requires a sufficiently sized dataset that depends on the complexity of the ML model. Generating such a large dataset from MD simulations is not ideal because of their high computation cost. In this study, we present a simple supervised ML method to predict the transport properties of materials. To simplify the model, an unsupervised ML method obtains an efficient representation of molecular movements. This method was applied to predict the viscosity of lubricant molecules in confinement with shear flow. Furthermore, simplicity facilitates the interpretation of the model to understand the molecular mechanics of viscosity. We revealed two types of molecular mechanisms that contribute to low viscosity.
MD-GAN with multi-particle input: the machine learning of long-time molecular behavior from short-time MD data
Feb 02, 2022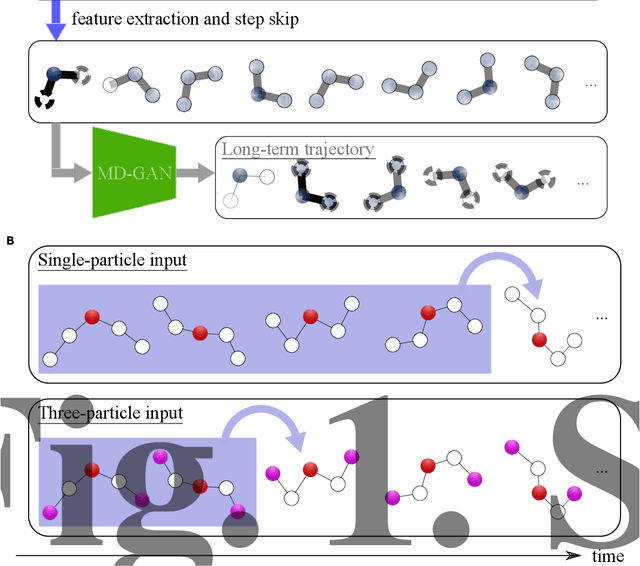
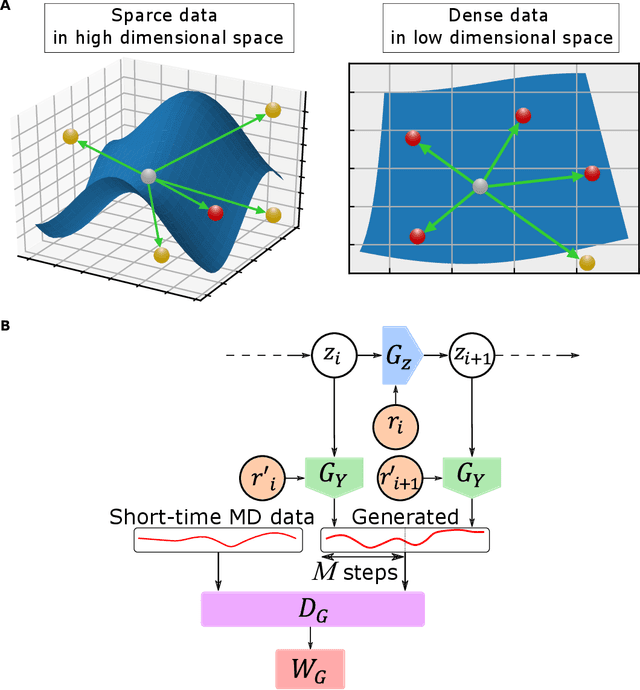
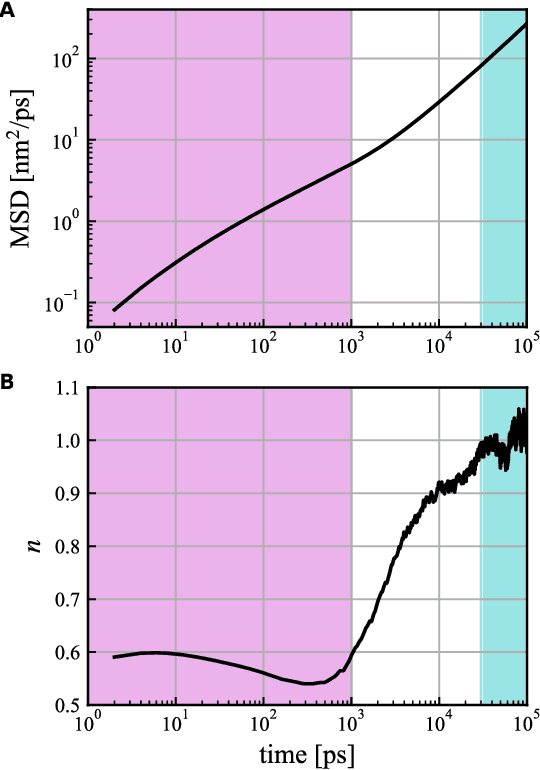
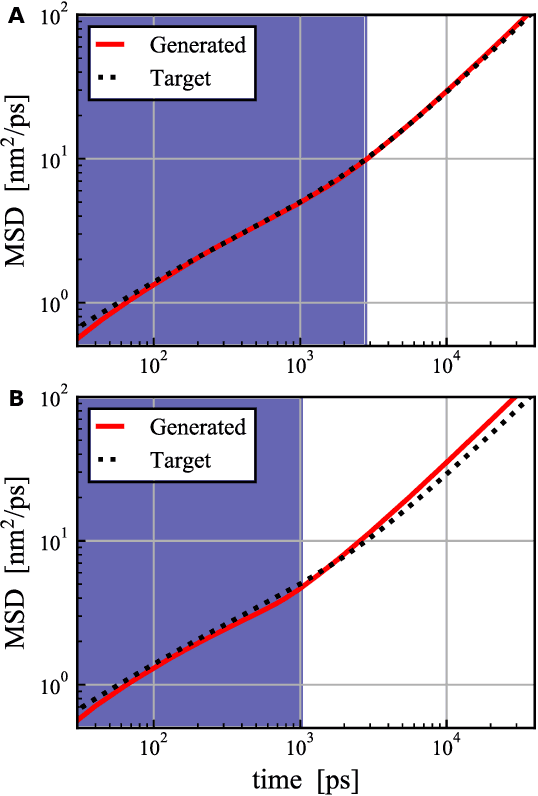
MD-GAN is a machine learning-based method that can evolve part of the system at any time step, accelerating the generation of molecular dynamics data. For the accurate prediction of MD-GAN, sufficient information on the dynamics of a part of the system should be included with the training data. Therefore, the selection of the part of the system is important for efficient learning. In a previous study, only one particle (or vector) of each molecule was extracted as part of the system. Therefore, we investigated the effectiveness of adding information from other particles to the learning process. In the experiment of the polyethylene system, when the dynamics of three particles of each molecule were used, the diffusion was successfully predicted using one-third of the time length of the training data, compared to the single-particle input. Surprisingly, the unobserved transition of diffusion in the training data was also predicted using this method.