 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeCGRclust: Chaos Game Representation for Twin Contrastive Clustering of Unlabelled DNA Sequences
Jul 01, 2024This study proposes CGRclust, a novel combination of unsupervised twin contrastive clustering of Chaos Game Representations (CGR) of DNA sequences, with convolutional neural networks (CNNs). To the best of our knowledge, CGRclust is the first method to use unsupervised learning for image classification (herein applied to two-dimensional CGR images) for clustering datasets of DNA sequences. CGRclust overcomes the limitations of traditional sequence classification methods by leveraging unsupervised twin contrastive learning to detect distinctive sequence patterns, without requiring DNA sequence alignment or biological/taxonomic labels. CGRclust accurately clustered twenty-five diverse datasets, with sequence lengths ranging from 664 bp to 100 kbp, including mitochondrial genomes of fish, fungi, and protists, as well as viral whole genome assemblies and synthetic DNA sequences. Compared with three recent clustering methods for DNA sequences (DeLUCS, iDeLUCS, and MeShClust v3.0.), CGRclust is the only method that surpasses 81.70% accuracy across all four taxonomic levels tested for mitochondrial DNA genomes of fish. Moreover, CGRclust also consistently demonstrates superior performance across all the viral genomic datasets. The high clustering accuracy of CGRclust on these twenty-five datasets, which vary significantly in terms of sequence length, number of genomes, number of clusters, and level of taxonomy, demonstrates its robustness, scalability, and versatility.
Map of Life: Measuring and Visualizing Species' Relatedness with "Molecular Distance Maps"
Jul 14, 2013
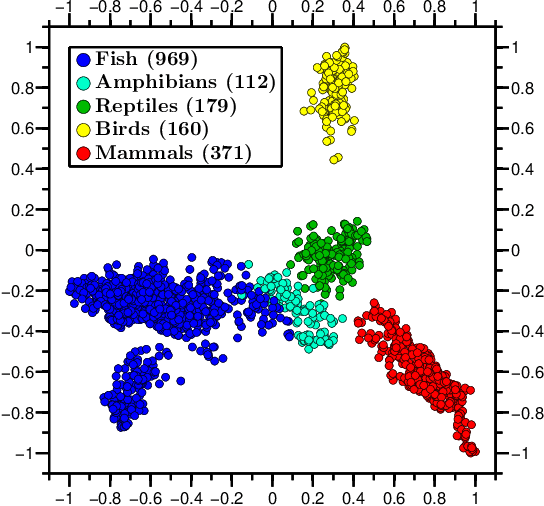
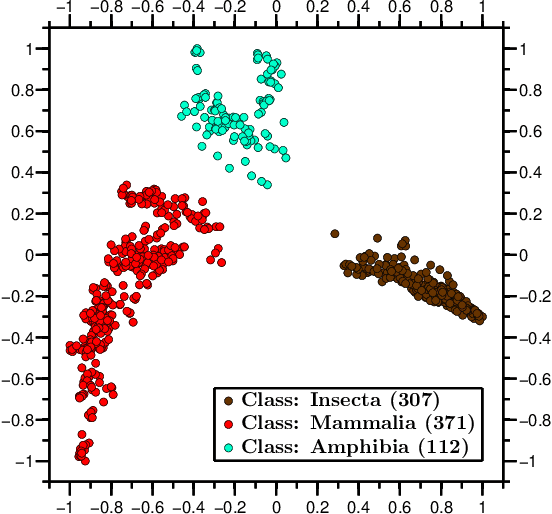
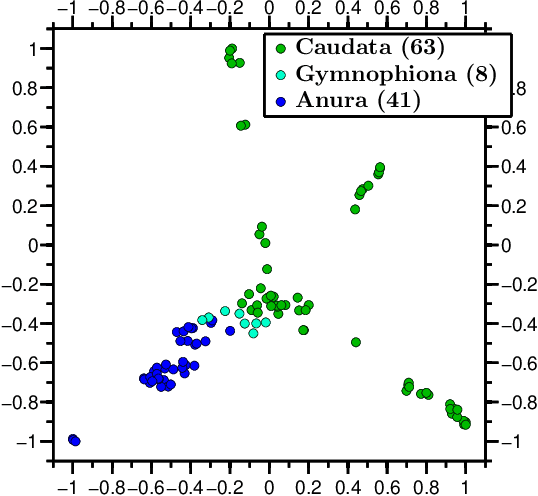
We propose a novel combination of methods that (i) portrays quantitative characteristics of a DNA sequence as an image, (ii) computes distances between these images, and (iii) uses these distances to output a map wherein each sequence is a point in a common Euclidean space. In the resulting "Molecular Distance Map" each point signifies a DNA sequence, and the geometric distance between any two points reflects the degree of relatedness between the corresponding sequences and species. Molecular Distance Maps present compelling visual representations of relationships between species and could be used for taxonomic clarifications, for species identification, and for studies of evolutionary history. One of the advantages of this method is its general applicability since, as sequence alignment is not required, the DNA sequences chosen for comparison can be completely different regions in different genomes. In fact, this method can be used to compare any two DNA sequences. For example, in our dataset of 3,176 mitochondrial DNA sequences, it correctly finds the mtDNA sequences most closely related to that of the anatomically modern human (the Neanderthal, the Denisovan, and the chimp), and it finds that the sequence most different from it belongs to a cucumber. Furthermore, our method can be used to compare real sequences to artificial, computer-generated, DNA sequences. For example, it is used to determine that the distances between a Homo sapiens sapiens mtDNA and artificial sequences of the same length and same trinucleotide frequencies can be larger than the distance between the same human mtDNA and the mtDNA of a fruit-fly. We demonstrate this method's promising potential for taxonomical clarifications by applying it to a diverse variety of cases that have been historically controversial, such as the genus Polypterus, the family Tarsiidae, and the vast (super)kingdom Protista.