 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgePredicting Biomedical Interactions with Higher-Order Graph Convolutional Networks
Oct 16, 2020
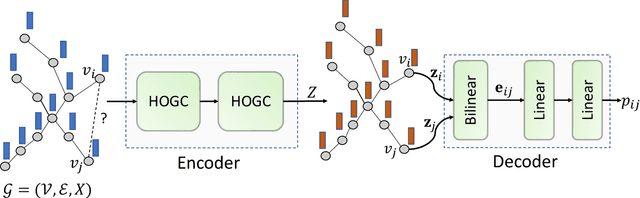
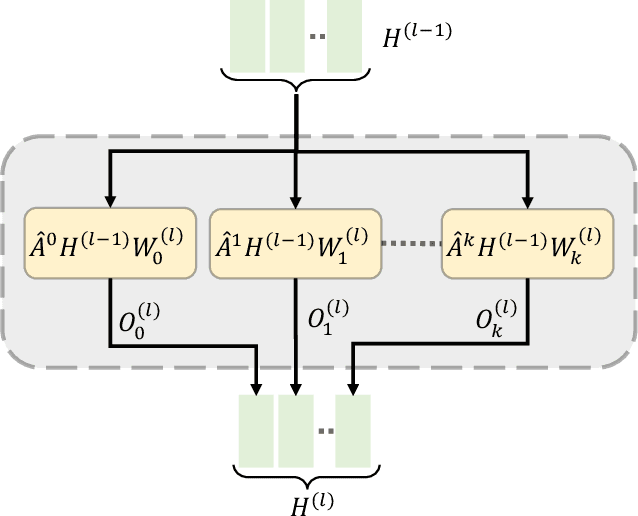
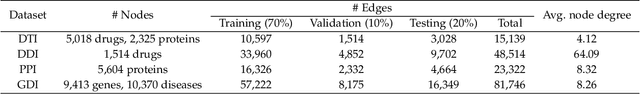
Biomedical interaction networks have incredible potential to be useful in the prediction of biologically meaningful interactions, identification of network biomarkers of disease, and the discovery of putative drug targets. Recently, graph neural networks have been proposed to effectively learn representations for biomedical entities and achieved state-of-the-art results in biomedical interaction prediction. These methods only consider information from immediate neighbors but cannot learn a general mixing of features from neighbors at various distances. In this paper, we present a higher-order graph convolutional network (HOGCN) to aggregate information from the higher-order neighborhood for biomedical interaction prediction. Specifically, HOGCN collects feature representations of neighbors at various distances and learns their linear mixing to obtain informative representations of biomedical entities. Experiments on four interaction networks, including protein-protein, drug-drug, drug-target, and gene-disease interactions, show that HOGCN achieves more accurate and calibrated predictions. HOGCN performs well on noisy, sparse interaction networks when feature representations of neighbors at various distances are considered. Moreover, a set of novel interaction predictions are validated by literature-based case studies.
Interpretable Structured Learning with Sparse Gated Sequence Encoder for Protein-Protein Interaction Prediction
Oct 16, 2020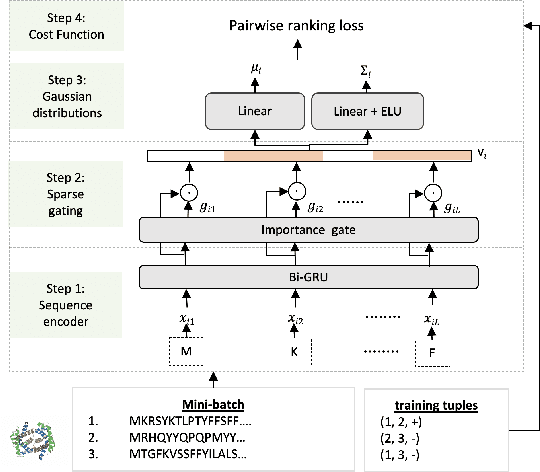
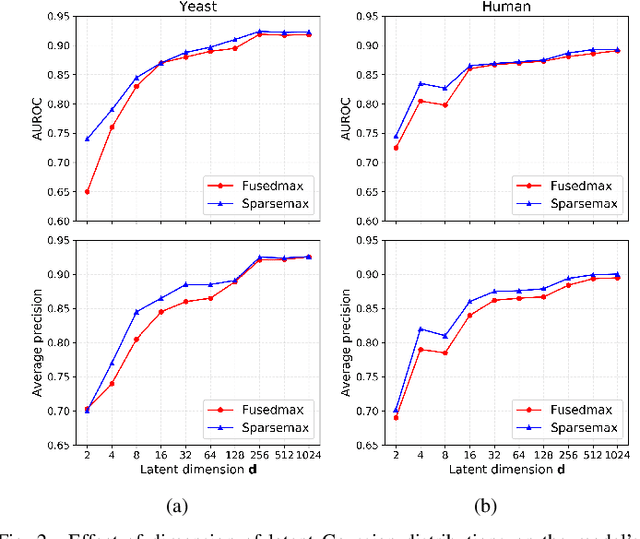
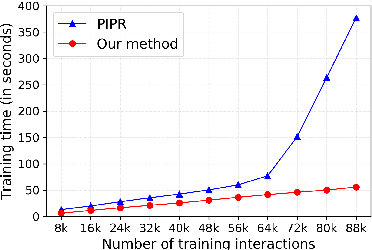

Predicting protein-protein interactions (PPIs) by learning informative representations from amino acid sequences is a challenging yet important problem in biology. Although various deep learning models in Siamese architecture have been proposed to model PPIs from sequences, these methods are computationally expensive for a large number of PPIs due to the pairwise encoding process. Furthermore, these methods are difficult to interpret because of non-intuitive mappings from protein sequences to their sequence representation. To address these challenges, we present a novel deep framework to model and predict PPIs from sequence alone. Our model incorporates a bidirectional gated recurrent unit to learn sequence representations by leveraging contextualized and sequential information from sequences. We further employ a sparse regularization to model long-range dependencies between amino acids and to select important amino acids (protein motifs), thus enhancing interpretability. Besides, the novel design of the encoding process makes our model computationally efficient and scalable to an increasing number of interactions. Experimental results on up-to-date interaction datasets demonstrate that our model achieves superior performance compared to other state-of-the-art methods. Literature-based case studies illustrate the ability of our model to provide biological insights to interpret the predictions.