 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeRNA Alternative Splicing Prediction with Discrete Compositional Energy Network
Mar 07, 2021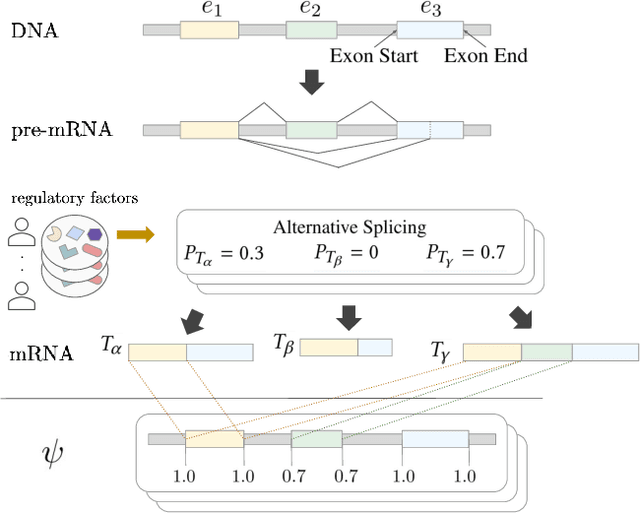
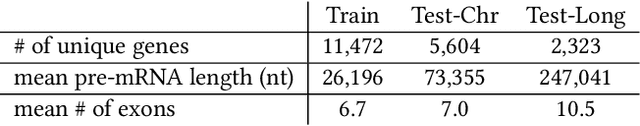
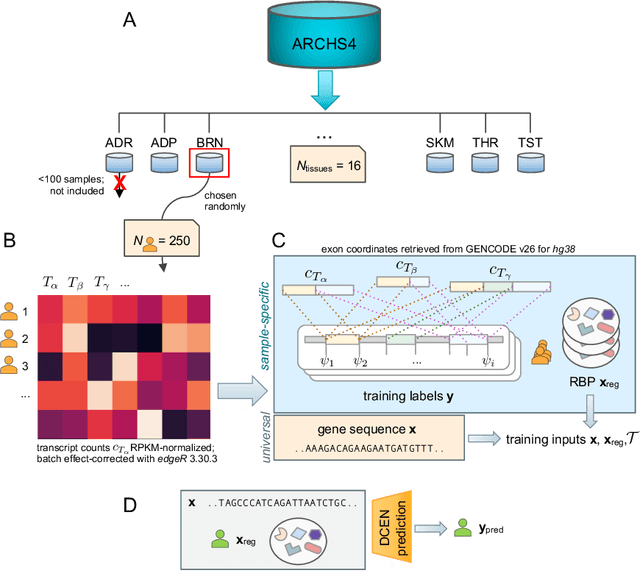
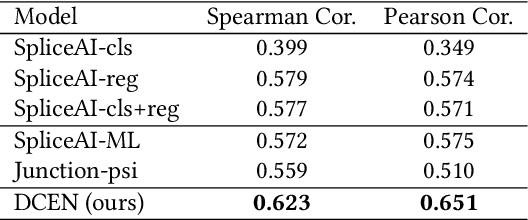
A single gene can encode for different protein versions through a process called alternative splicing. Since proteins play major roles in cellular functions, aberrant splicing profiles can result in a variety of diseases, including cancers. Alternative splicing is determined by the gene's primary sequence and other regulatory factors such as RNA-binding protein levels. With these as input, we formulate the prediction of RNA splicing as a regression task and build a new training dataset (CAPD) to benchmark learned models. We propose discrete compositional energy network (DCEN) which leverages the hierarchical relationships between splice sites, junctions and transcripts to approach this task. In the case of alternative splicing prediction, DCEN models mRNA transcript probabilities through its constituent splice junctions' energy values. These transcript probabilities are subsequently mapped to relative abundance values of key nucleotides and trained with ground-truth experimental measurements. Through our experiments on CAPD, we show that DCEN outperforms baselines and ablation variants.