 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeUnderstanding Anharmonic Effects on Hydrogen Desorption Characteristics of Mg$_n$H$_{2n}$ Nanoclusters by ab initio trained Deep Neural Network
Paper and Code
Nov 27, 2021
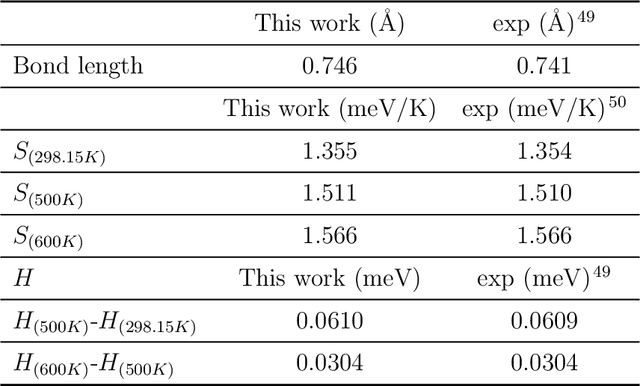

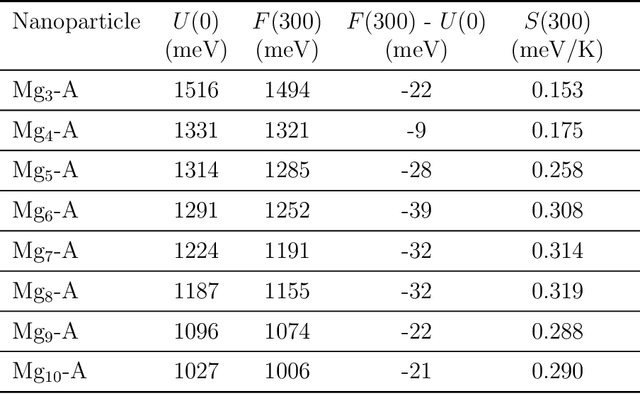
Magnesium hydride (MgH$_2$) has been widely studied for effective hydrogen storage. However, its bulk desorption temperature (553 K) is deemed too high for practical applications. Besides doping, a strategy to decrease such reaction energy for releasing hydrogen is the use of MgH$_2$-based nanoparticles (NPs). Here, we investigate first the thermodynamic properties of Mg$_n$H$_{2n}$ NPs ($n<10$) from first-principles, in particular by assessing the anharmonic effects on the enthalpy, entropy and thermal expansion by means of the Stochastic Self Consistent Harmonic Approximation (SSCHA). The latter method goes beyond previous approaches, typically based on molecular mechanics and the quasi-harmonic approximation, allowing the ab initio calculation of the fully-anharmonic free energy. We find an almost linear dependence on temperature of the interatomic bond lengths - with a relative variation of few percent over 300K -, alongside with a bond distance decrease of the Mg-H bonds. In order to increase the size of NPs toward experiments of hydrogen desorption from MgH$_2$ we devise a computationally effective Machine Learning model trained to accurately determine the forces and total energies (i.e. the potential energy surfaces), integrating the latter with the SSCHA model to fully include the anharmonic effects. We find a significative decrease of the H-desorption temperature for sub-nanometric clusters Mg$_n$H$_{2n}$ with $n \leq 10$, with a non-negligible, although little effect due to anharmonicities (up to 10%).