 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeParticle Swarm Based Hyper-Parameter Optimization for Machine Learned Interatomic Potentials
Paper and Code
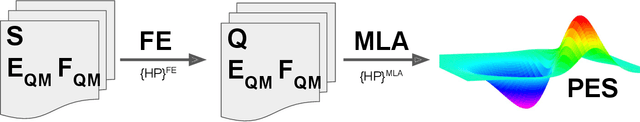
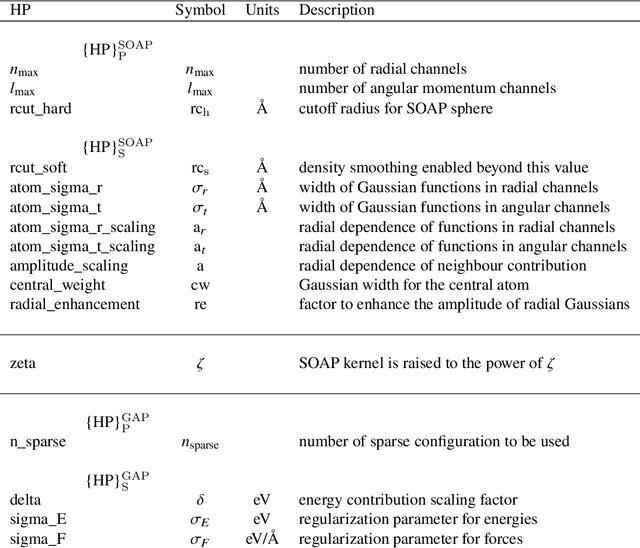
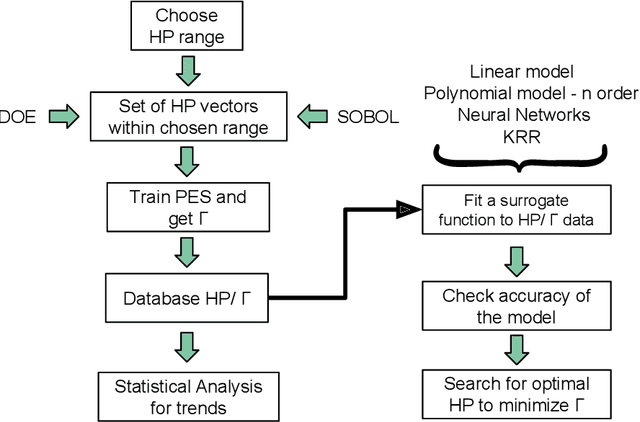
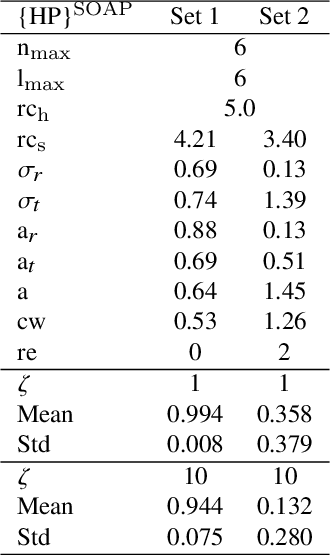
Modeling non-empirical and highly flexible interatomic potential energy surfaces (PES) using machine learning (ML) approaches is becoming popular in molecular and materials research. Training an ML-PES is typically performed in two stages: feature extraction and structure-property relationship modeling. The feature extraction stage transforms atomic positions into a symmetry-invariant mathematical representation. This representation can be fine-tuned by adjusting on a set of so-called "hyper-parameters" (HPs). Subsequently, an ML algorithm such as neural networks or Gaussian process regression (GPR) is used to model the structure-PES relationship based on another set of HPs. Choosing optimal values for the two sets of HPs is critical to ensure the high quality of the resulting ML-PES model. In this paper, we explore HP optimization strategies tailored for ML-PES generation using a custom-coded parallel particle swarm optimizer (available freely at https://github.com/suresh0807/PPSO.git). We employ the smooth overlap of atomic positions (SOAP) descriptor in combination with GPR-based Gaussian approximation potentials (GAP) and optimize HPs for four distinct systems: a toy C dimer, amorphous carbon, $\alpha$-Fe, and small organic molecules (QM9 dataset). We propose a two-step optimization strategy in which the HPs related to the feature extraction stage are optimized first, followed by the optimization of the HPs in the training stage. This strategy is computationally more efficient than optimizing all HPs at the same time by means of significantly reducing the number of ML models needed to be trained to obtain the optimal HPs. This approach can be trivially extended to other combinations of descriptor and ML algorithm and brings us another step closer to fully automated ML-PES generation.