 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeHigh throughput screening with machine learning
Paper and Code
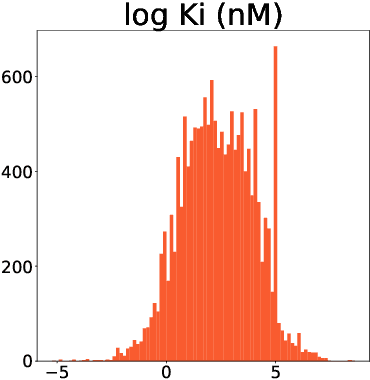
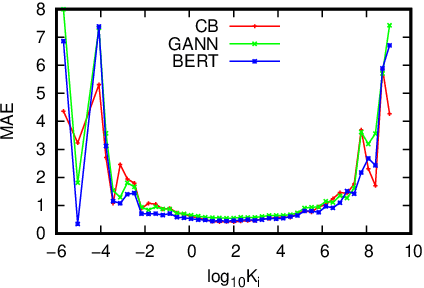
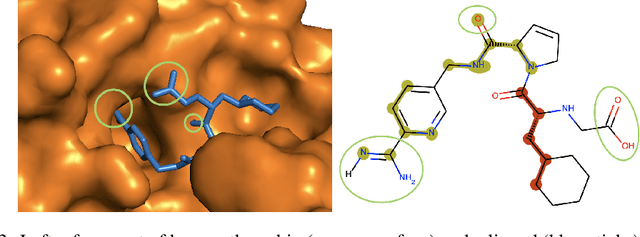
This study assesses the efficiency of several popular machine learning approaches in the prediction of molecular binding affinity: CatBoost, Graph Attention Neural Network, and Bidirectional Encoder Representations from Transformers. The models were trained to predict binding affinities in terms of inhibition constants $K_i$ for pairs of proteins and small organic molecules. First two approaches use thoroughly selected physico-chemical features, while the third one is based on textual molecular representations - it is one of the first attempts to apply Transformer-based predictors for the binding affinity. We also discuss the visualization of attention layers within the Transformer approach in order to highlight the molecular sites responsible for interactions. All approaches are free from atomic spatial coordinates thus avoiding bias from known structures and being able to generalize for compounds with unknown conformations. The achieved accuracy for all suggested approaches prove their potential in high throughput screening.