 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeEquiCPI: SE(3)-Equivariant Geometric Deep Learning for Structure-Aware Prediction of Compound-Protein Interactions
Paper and Code


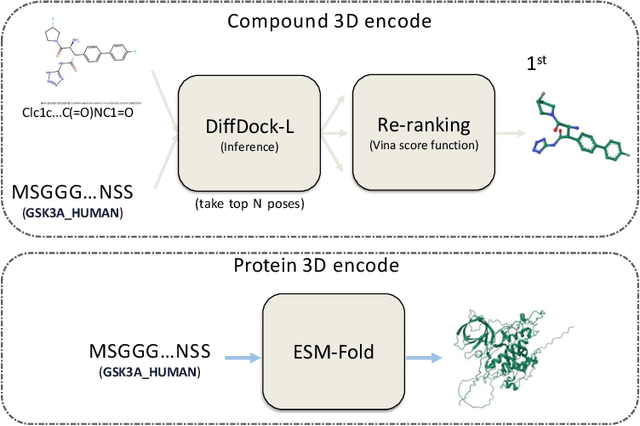

Accurate prediction of compound-protein interactions (CPI) remains a cornerstone challenge in computational drug discovery. While existing sequence-based approaches leverage molecular fingerprints or graph representations, they critically overlook three-dimensional (3D) structural determinants of binding affinity. To bridge this gap, we present EquiCPI, an end-to-end geometric deep learning framework that synergizes first-principles structural modeling with SE(3)-equivariant neural networks. Our pipeline transforms raw sequences into 3D atomic coordinates via ESMFold for proteins and DiffDock-L for ligands, followed by physics-guided conformer re-ranking and equivariant feature learning. At its core, EquiCPI employs SE(3)-equivariant message passing over atomic point clouds, preserving symmetry under rotations, translations, and reflections, while hierarchically encoding local interaction patterns through tensor products of spherical harmonics. The proposed model is evaluated on BindingDB (affinity prediction) and DUD-E (virtual screening), EquiCPI achieves performance on par with or exceeding the state-of-the-art deep learning competitors.