 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeAUGUR, A flexible and efficient optimization algorithm for identification of optimal adsorption sites
Paper and Code
Sep 24, 2024

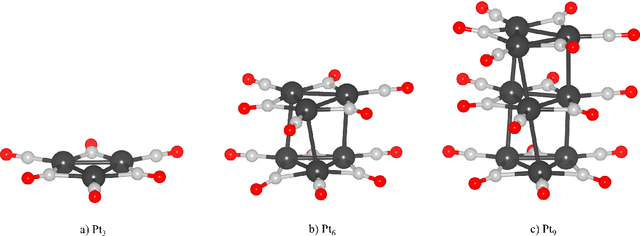
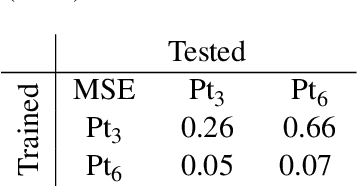
In this paper, we propose a novel flexible optimization pipeline for determining the optimal adsorption sites, named AUGUR (Aware of Uncertainty Graph Unit Regression). Our model combines graph neural networks and Gaussian processes to create a flexible, efficient, symmetry-aware, translation, and rotation-invariant predictor with inbuilt uncertainty quantification. This predictor is then used as a surrogate for a data-efficient Bayesian Optimization scheme to determine the optimal adsorption positions. This pipeline determines the optimal position of large and complicated clusters with far fewer iterations than current state-of-the-art approaches. Further, it does not rely on hand-crafted features and can be seamlessly employed on any molecule without any alterations. Additionally, the pooling properties of graphs allow for the processing of molecules of different sizes by the same model. This allows the energy prediction of computationally demanding systems by a model trained on comparatively smaller and less expensive ones