Get our free extension to see links to code for papers anywhere online!Free add-on: code for papers everywhere!Free add-on: See code for papers anywhere!
 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeAtomistic Simulations for Reactions and Spectroscopy in the Era of Machine Learning -- Quo Vadis?
Paper and Code
Jan 11, 2022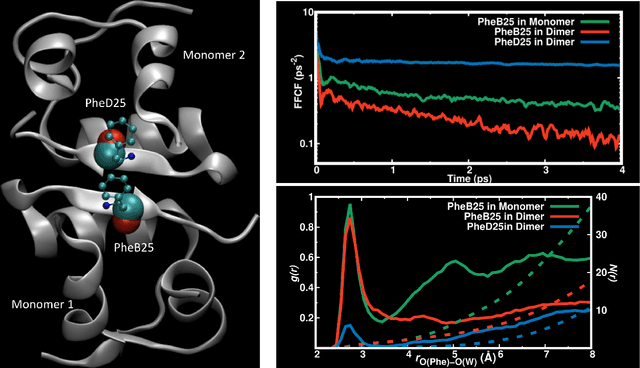

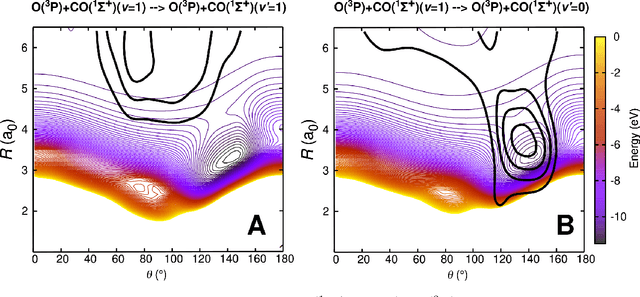
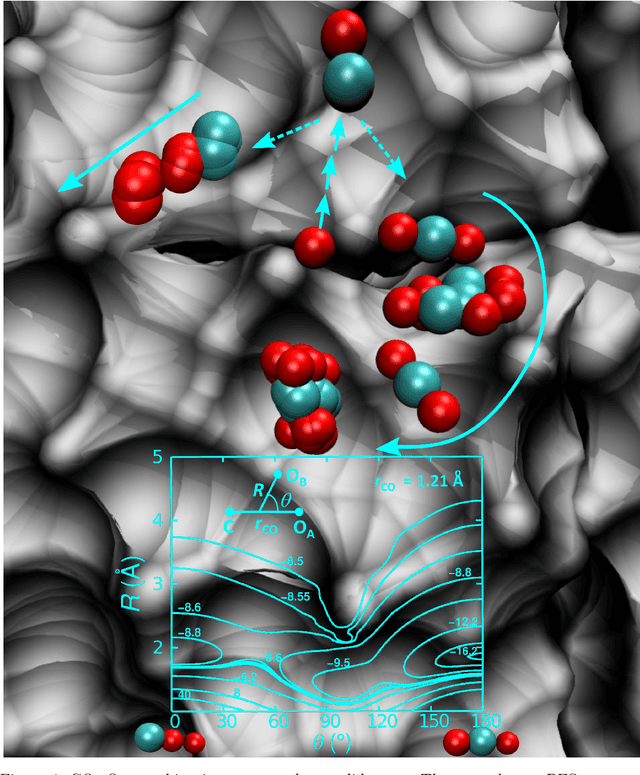
Atomistic simulations using accurate energy functions can provide molecular-level insight into functional motions of molecules in the gas- and in the condensed phase. Together with recently developed and currently pursued efforts in integrating and combining this with machine learning techniques provides a unique opportunity to bring such dynamics simulations closer to reality. This perspective delineates the present status of the field from efforts of others in the field and some of your own work and discusses open questions and future prospects.
View paper on