 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeAPObind: A Dataset of Ligand Unbound Protein Conformations for Machine Learning Applications in De Novo Drug Design
Paper and Code
Aug 25, 2021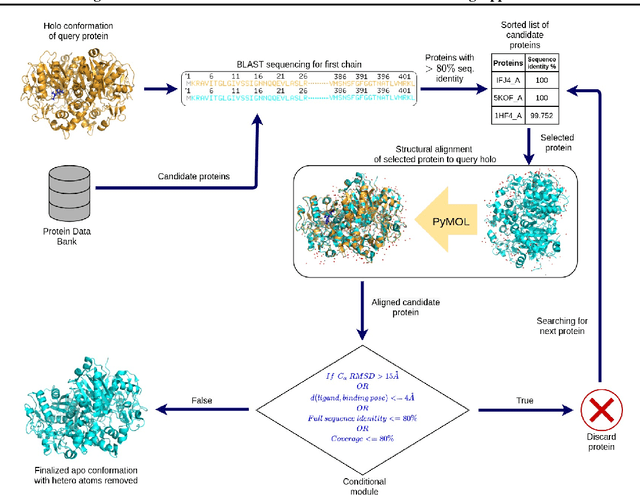

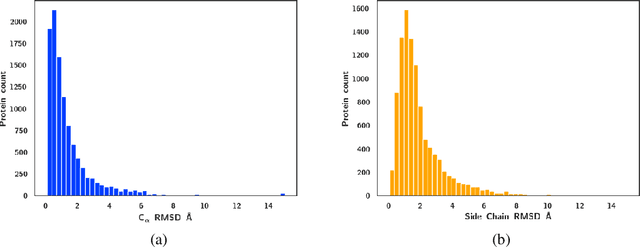
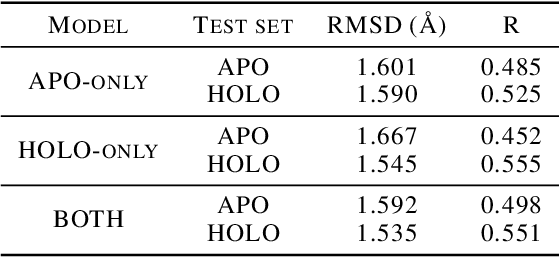
Protein-ligand complex structures have been utilised to design benchmark machine learning methods that perform important tasks related to drug design such as receptor binding site detection, small molecule docking and binding affinity prediction. However, these methods are usually trained on only ligand bound (or holo) conformations of the protein and therefore are not guaranteed to perform well when the protein structure is in its native unbound conformation (or apo), which is usually the conformation available for a newly identified receptor. A primary reason for this is that the local structure of the binding site usually changes upon ligand binding. To facilitate solutions for this problem, we propose a dataset called APObind that aims to provide apo conformations of proteins present in the PDBbind dataset, a popular dataset used in drug design. Furthermore, we explore the performance of methods specific to three use cases on this dataset, through which, the importance of validating them on the APObind dataset is demonstrated.