 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeAlgorithmic Bio-surveillance For Precise Spatio-temporal Prediction of Zoonotic Emergence
Paper and Code
Jan 23, 2018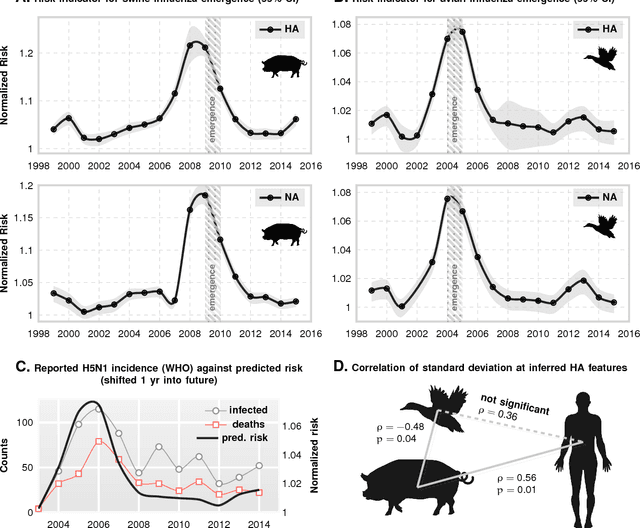


Viral zoonoses have emerged as the key drivers of recent pandemics. Human infection by zoonotic viruses are either spillover events -- isolated infections that fail to cause a widespread contagion -- or species jumps, where successful adaptation to the new host leads to a pandemic. Despite expensive bio-surveillance efforts, historically emergence response has been reactive, and post-hoc. Here we use machine inference to demonstrate a high accuracy predictive bio-surveillance capability, designed to pro-actively localize an impending species jump via automated interrogation of massive sequence databases of viral proteins. Our results suggest that a jump might not purely be the result of an isolated unfortunate cross-infection localized in space and time; there are subtle yet detectable patterns of genotypic changes accumulating in the global viral population leading up to emergence. Using tens of thousands of protein sequences simultaneously, we train models that track maximum achievable accuracy for disambiguating host tropism from the primary structure of surface proteins, and show that the inverse classification accuracy is a quantitative indicator of jump risk. We validate our claim in the context of the 2009 swine flu outbreak, and the 2004 emergence of H5N1 subspecies of Influenza A from avian reservoirs; illustrating that interrogation of the global viral population can unambiguously track a near monotonic risk elevation over several preceding years leading to eventual emergence.