 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeA2I Transformer: Permutation-equivariant attention network for pairwise and many-body interactions with minimal featurization
Paper and Code
Oct 27, 2021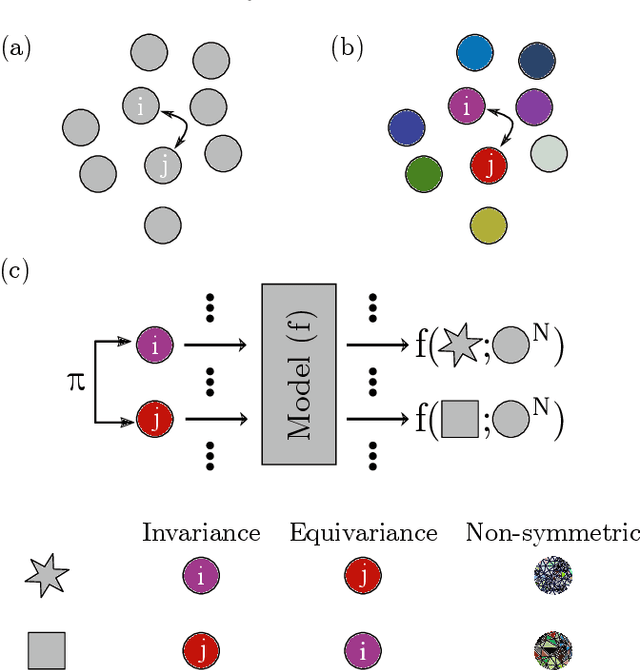
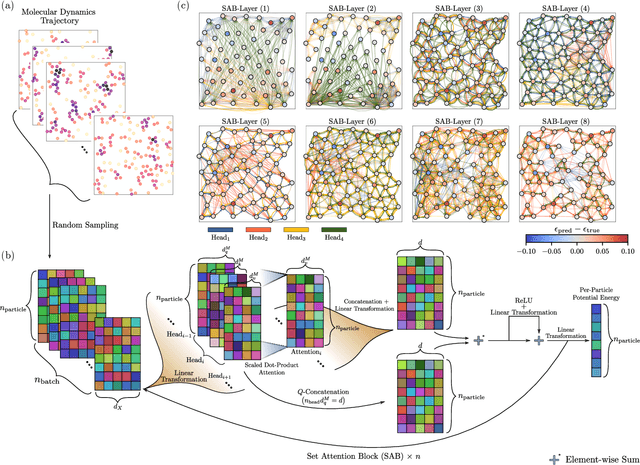
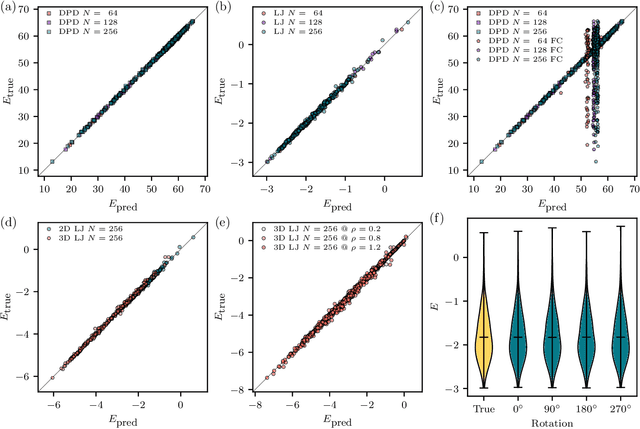
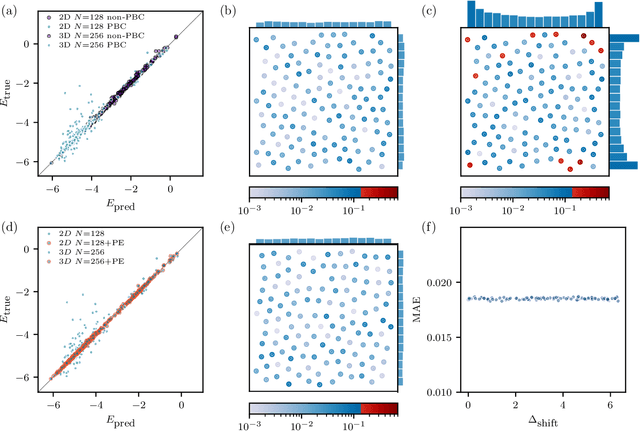
The combination of neural network potential (NNP) with molecular simulations plays an important role in an efficient and thorough understanding of a molecular system's potential energy surface (PES). However, grasping the interplay between input features and their local contribution to NNP is growingly evasive due to heavy featurization. In this work, we suggest an end-to-end model which directly predicts per-atom energy from the coordinates of particles, avoiding expert-guided featurization of the network input. Employing self-attention as the main workhorse, our model is intrinsically equivariant under the permutation operation, resulting in the invariance of the total potential energy. We tested our model against several challenges in molecular simulation problems, including periodic boundary condition (PBC), $n$-body interaction, and binary composition. Our model yielded stable predictions in all tested systems with errors significantly smaller than the potential energy fluctuation acquired from molecular dynamics simulations. Thus, our work provides a minimal baseline model that encodes complex interactions in a condensed phase system to facilitate the data-driven analysis of physicochemical systems.